4 Pseudopotential Explanation
DS-PAW currently supports three formats of pseudopotentials: .paw, .potcar, and .pawpsp. Users can specify the pseudopotential type using the sys.pseudoType parameter.
4.1 hzw internal PAW pseudopotential
The DS-PAW defaults to using the hzw pseudopotentials (.paw), with the corresponding parameter sys.pseudoType set to -1. In this case, DS-PAW will read the pseudopotential files from the installation path /pseudopotential. Currently, the hzw pseudopotential library contains 72 elements, covering elements 1-86 in the periodic table (lanthanides are currently supported only up to Lanthanum).
Regarding the accuracy of the hzw pseudopotential: the quick start guide and application examples perform calculations based on multiple functionalities, and the results are in good agreement with the literature, which validates that the hzw pseudopotential exhibits high accuracy in various functional calculations.
Furthermore, for the 72 elements in the pseudopotential library, calculations of equation of state fitting to obtain the equilibrium cell volume were performed based on the 1.0 version pseudopotentials for the corresponding elemental solids. The test objects included the 72 elements’ LDA and PBE functional pseudopotentials, totaling 144 pseudopotential files. The calculated volumes were compared with those obtained from the quantum chemistry software WIEN2k, and the calculation errors are displayed in the periodic table as follows:
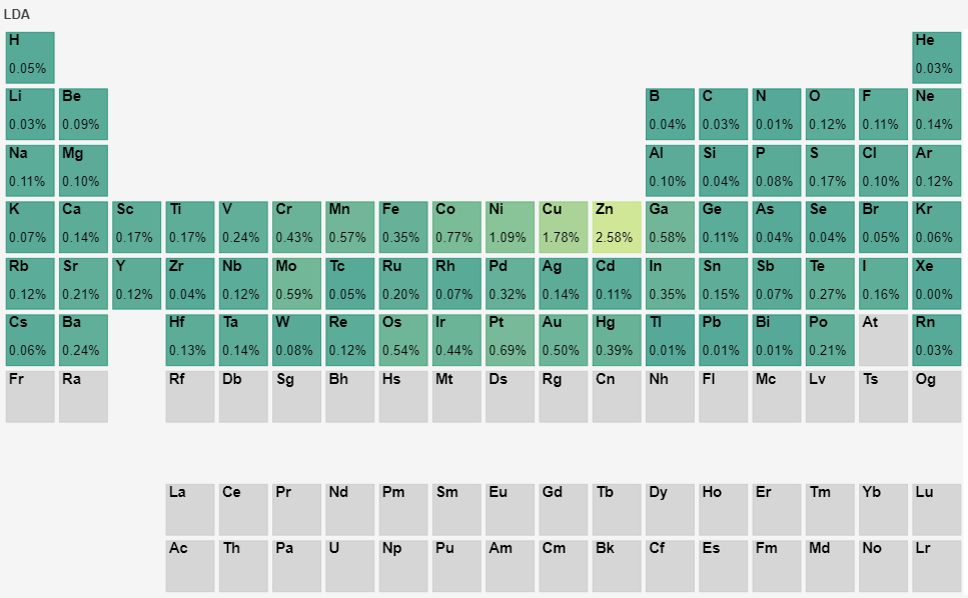
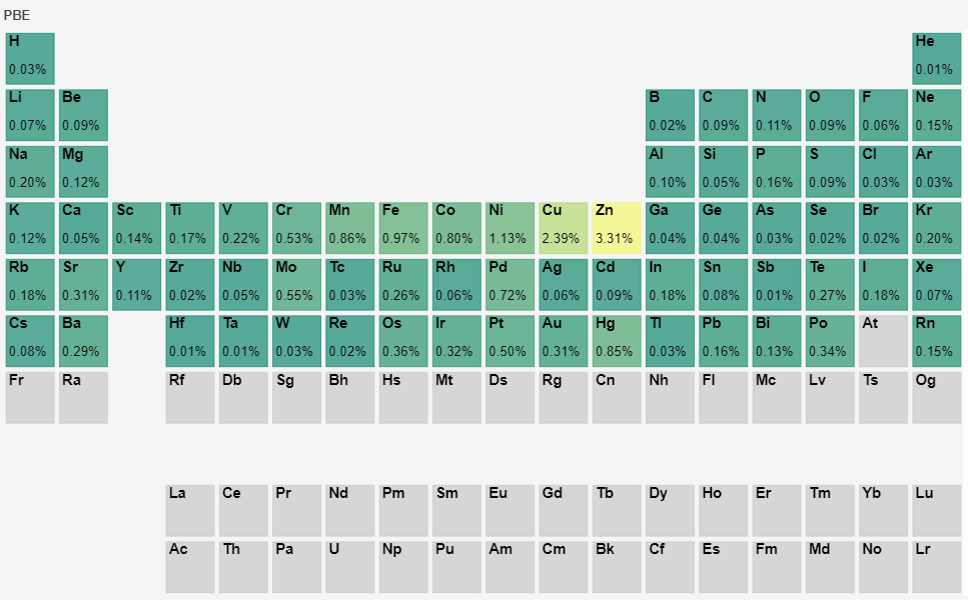
By comparison, the equilibrium volume obtained by fitting the equation of state in version 1.0 is basically consistent with the results from WIEN2k software. The largest errors are for the element Zn, with errors of 2.58% and 3.31% for the LDA and PBE pseudopotentials, respectively. Optimization of the pseudopotentials for these two elements is underway. The errors for the remaining elements are basically controlled within 0.1%, which further validates the overall accuracy of the pseudopotential library.
The 1.1 version of the pseudopotentials will be released on 2024/12/31. Performance data will be announced shortly, stay tuned…
4.2 VASP Pseudopotentials
DS-PAW provides an interface for using external POTCAR formatted pseudopotentials (.potcar), with the corresponding parameter sys.pseudoType set to 10. In this case, DS-PAW will read the pseudopotential files from the default path ./. Due to copyright restrictions, DS-PAW only provides the interface for using VASP pseudopotentials, and the pseudopotential files must be prepared by the user.
When using this feature, users need to modify the pseudopotential filenames accordingly. For example, if using the LDA pseudopotential for silicon, the corresponding POTCAR should be renamed to Si_LDA.potcar and placed in the specified directory (which can be set via sys.pseudoPath).
4.3 GBRV Pseudopotential
DS-PAW provides an interface for using external gbrv-formatted pseudopotentials (.pawpsp), with the corresponding parameter sys.pseudoType set to 11. In this case, DS-PAW will read the pseudopotential files from the default path ./. The gbrv pseudopotential library is a freely available set of pseudopotentials, offering files for a total of 64 elements. The download website is http://www.physics.rutgers.edu/gbrv/. When downloading, please note that DS-PAW supports the PAW format for Abinit. When using these pseudopotentials, the user needs to rename the pseudopotential files accordingly. For example, if using the LDA pseudopotential for silicon, the corresponding pseudopotential file should be renamed to Si_LDA.pawpsp and placed in the specified directory (which can be set via sys.pseudoPath).
4.4 Compare Pseudopotentials
\(Si\) band structure calculation
To compare the computational results of the three pseudopotentials, this section uses silicon (Si) as an example. Band structure calculations were performed using all three pseudopotentials. The figure below shows a comparison of the band structures. This comparison demonstrates a high degree of consistency among the three pseudopotentials in describing the Si band structure, thus validating the accuracy of the hzw pseudopotential.
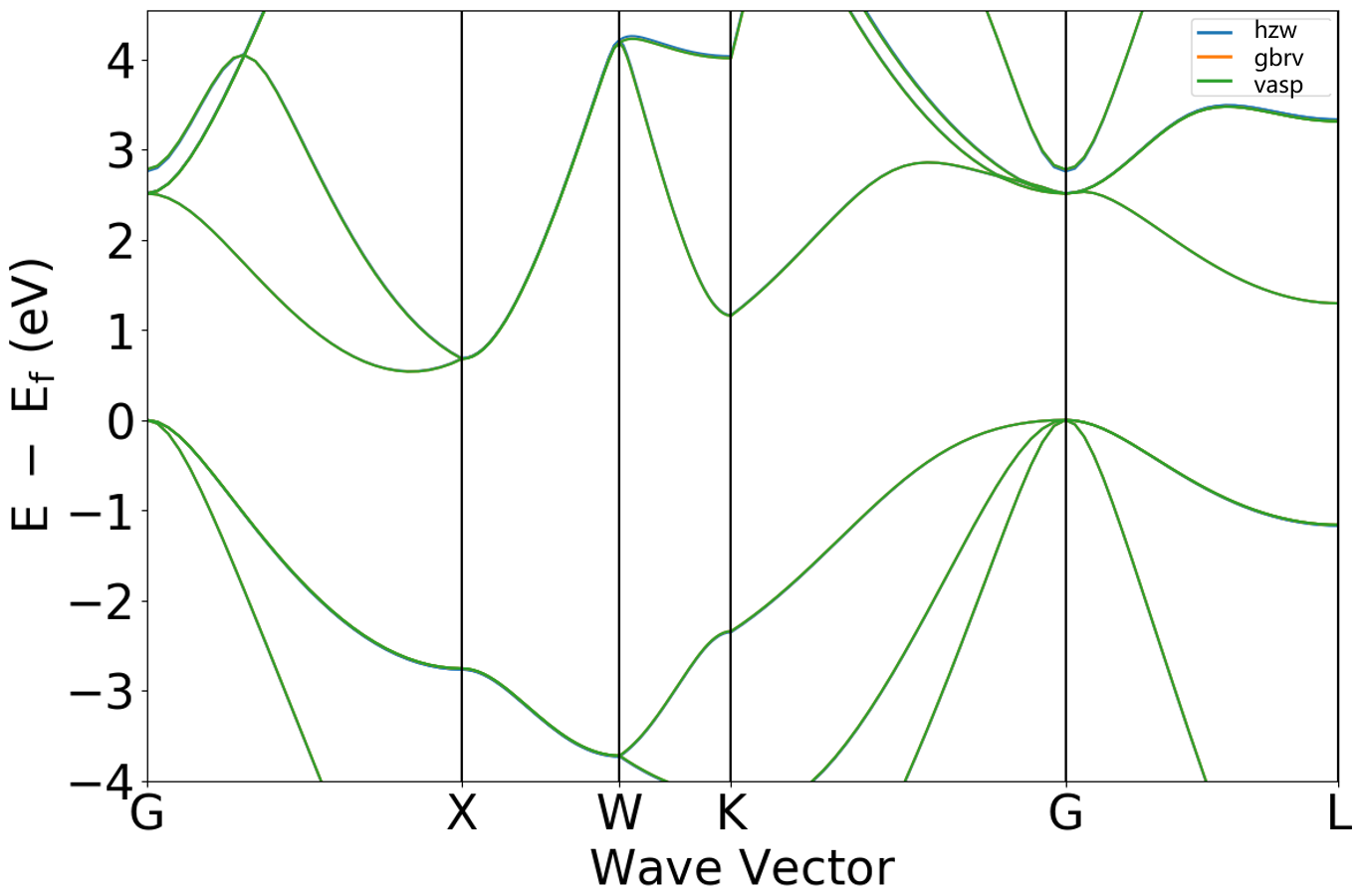
*Data Source: The data for this figure was provided by a collaborator of Hongzhiwei.
Multi-system band gap calculation
This section presents band gap calculations using HSE06 with version 1.0 pseudopotentials for multiple systems. The band gap values obtained are compared with those calculated using VASP and RESCU software, as reported in the literature, along with those from DS-PAW calculations. The resulting figure (shown below) further validates the accuracy of the hzw pseudopotentials.
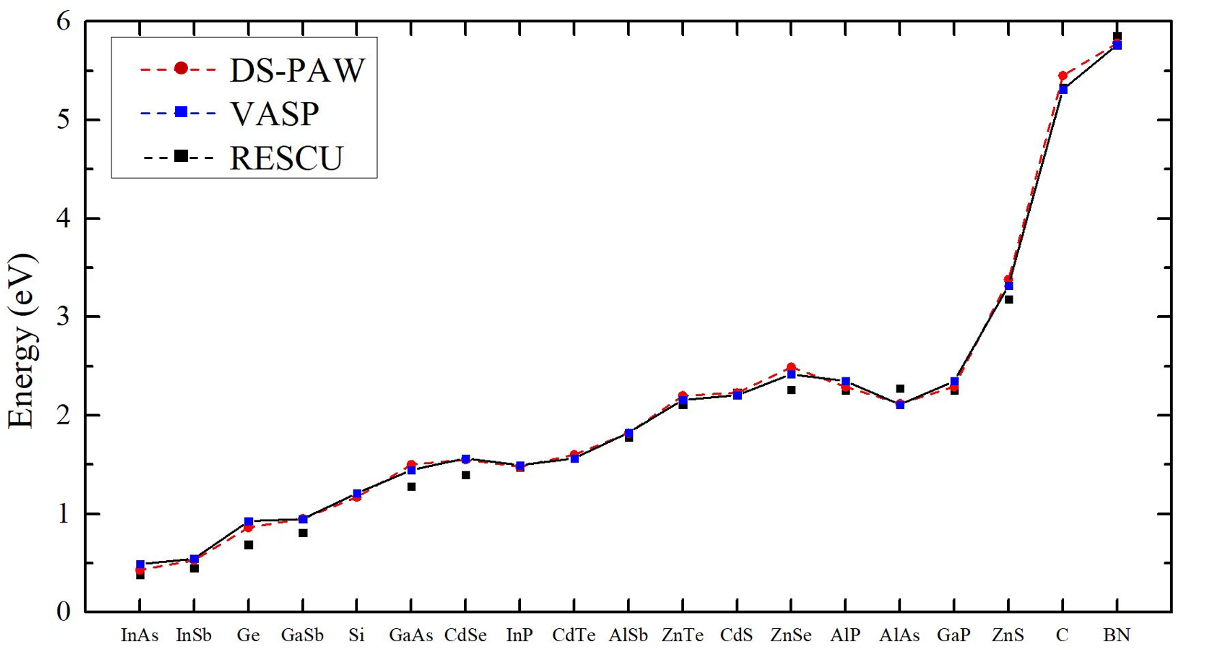
*Data Source: https://doi.org/10.1103/PhysRevB.97.075139